January 12, 2024, by Brigitte Nerlich
Sickle cell disease and gene editing
Before the end of 2023, I had rarely heard of sickle cell disease (or anaemia). I knew it existed, but that was all. Then, around November and December, it was suddenly in the media spotlight, because UK and US health authorities approved a new therapy and because the new therapy used the still new and shiny method of CRISPR-based gene editing.
This brought hope to many sufferers and it brought the disease to public attention. As Sharl Azar, medical director of the Comprehensive Sickle Cell Disease Treatment Center at Massachusetts General Hospital in Boston, pointed out, this is “allowing sickle cell to come to the social consciousness […] That’s a huge boon to the community”- as reported in USA Today on 8 December.
Reading this, I started to look a bit more closely at what sickle cell disease was, what gene editing promised to do, how the media reacted and what those who got or might get the new therapy think about it. The blog post I began to write got too long. So I have split it into two parts.
In this post I try to explain what the disease is, how science and medicine researched it, who is affected by it, and what the new CRISPR treatment involves – this is still quite long and a bit dry, but I hope it is informative. You can skip the parts you know. [I am not a medic. So, let me know if you find errors in my summaries]
In the other post, I’ll home in on patient stories as told to journalists, focussing in particular on the issue of identity and how the disease and the treatment shape and transform not only the body but also the self. I think it is really important to listen to patient voices (and probably to more than those quoted in the newspapers – but that would be a whole research project, continuing work carried out before the advent of CRISPR).
What is sickle cell disease?
“Sickle cell disease is an inherited blood disorder marked by defective hemoglobin [a protein in red blood cells that carries oxygen]. It inhibits the ability of hemoglobin in red blood cells to carry oxygen. Sickle cells tend to stick together, blocking small blood vessels causing painful and damaging complications.”
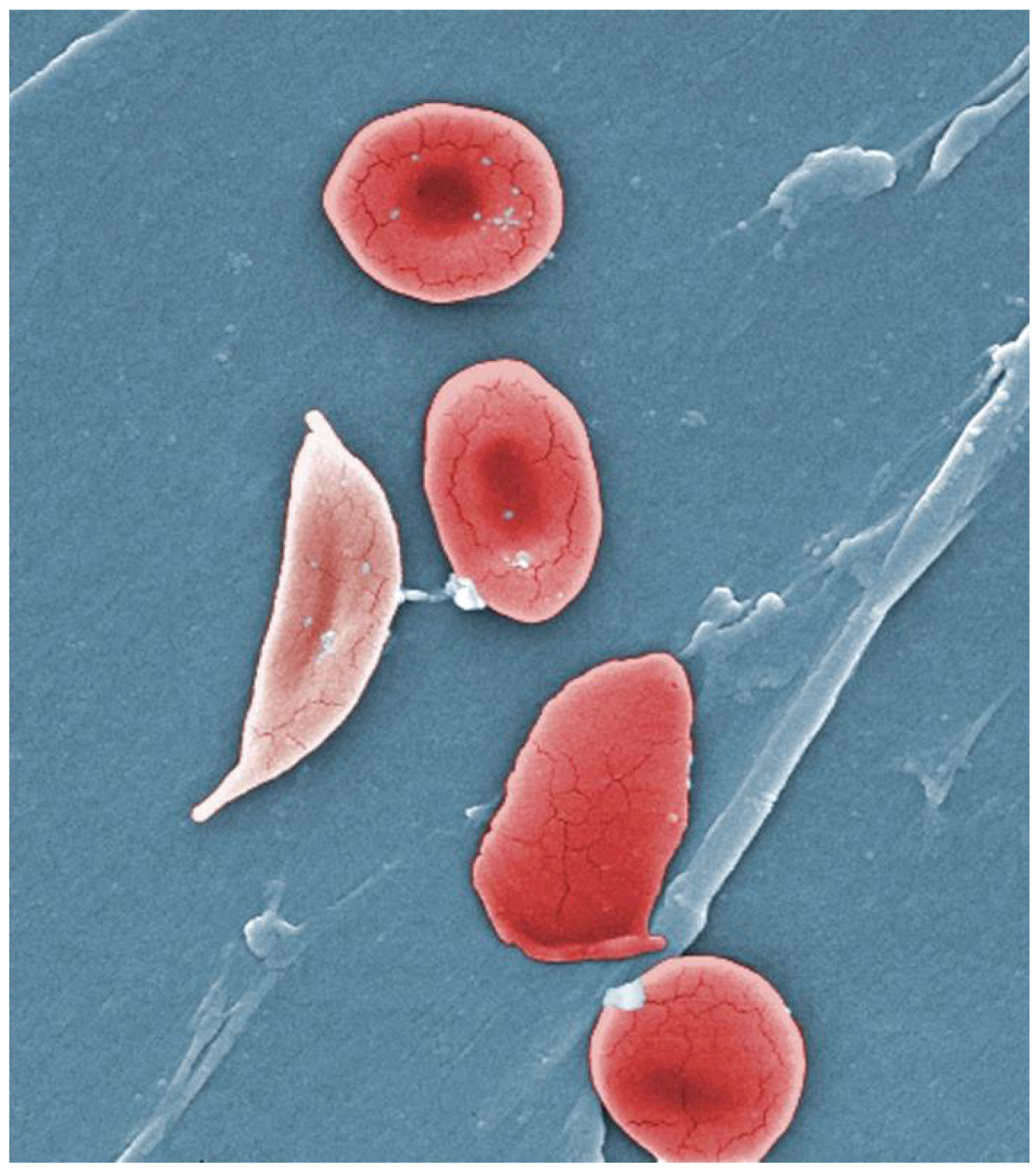
The disease gets its name from the shape of the defective blood cells: “Red blood cells with normal hemoglobin are smooth, disk-shaped, and flexible, like doughnuts without holes. They can move through the blood vessels easily. Cells with sickle cell hemoglobin are stiff and sticky. When they lose their oxygen, they form into the shape of a sickle or crescent, like the letter C.”
Who is affected?
Approximately 250 million people worldwide carry the gene for sickle cell disease, many living in Sub-Saharan Africa. About 100,000 people in the US are affected by sickle cell diseaseand about 17,500 in the UK.
As the UK Sickle Cell Society points out – and that surprised me: “Sickle Cell Disorder is the fastest growing condition in the UK, but is extremely underserved by treatment options.” ‘Underserved’, ‘underfunded’, ‘underused’ and many other such words describe the sickle cell situation rather well…and we have to see whether, how, where and for whom the new therapy changes this or not.
People affected by the disease experience episodes of intense pain and other serious symptoms. Treatments can involve frequent blood transfusions and stem cell transplants from a donor. Overall, the disease affects people’s lives severely.
Discoveries and treatments
Sickle cell disease was “first described in 1910 by noted Chicago-based internist James Bryan Herrick”. In the 1940s doctors noticed that patients who had sickle cell disease were more likely to survive malaria. For “parents who each carry the sickle cell trait, the chance that their child will also have the trait — and be immune to malaria — is 50 percent. There is a 25 percent chance that the child will have neither sickle cell anemia nor the trait which enables immunity to malaria. Finally, the chances that their child will have two copies of the gene, and therefore sickle cell anemia, is also 25 percent.”
In 1949, Linus Pauling, an American theoretical chemist, biochemist, and his colleagues published an article in Science on sickle cell anemia as a molecular disease. They showed that “that hemoglobin from patients suffering from sickle cell anemia has a different electrical charge than hemoglobin from healthy individuals. This result demonstrated for the first time that an abnormal protein could be causally linked to a disease, and that genes determined the structure of protein”. This opened up new ways of studying ‘molecular diseases’.
Pauling suggested that “… our structural chemistry and understanding of molecules is getting to the point where it should be of assistance in converting medicine into a real science”. (More history here!) I wonder what he’d think of developments today.
In 1972 “Congress passed the National Sickle Cell Disease Control Act which, for the first time, provided authority to establish education, information, screening, testing, counseling, research, and treatment programs.”
The medication Hydroxyurea (a chemotherapy medication) was approved for various diseases in 1967 and then for sickle cell in 1998, but it remained underused, although it has been approved in Ghana, for example, in 2018.
The disease can cause not only unbearable crises of pain but also, amongst other things, strokes. It was therefore a relief to sufferers when, in 1998, an ultrasound stroke screen was established, which, again, remained, however, underused.
In 2008 Vijay Sankaran and others wrote prophetically: “As a stage-specific component involved in the silencing of γ-globin expression, BCL11A, therefore, emerges as a new therapeutic target for reactivation of HbF in sickle cell disease and the β-thalassemias.” 15 years such a therapy was realised.
In 2019 the US Food and Drug Administration (FDA) approved Voxelotor, which is the first drug approved by the FDA for sickle cell disease based solely on data showing an increase in haemoglobin. It helps haemoglobin (a protein in red blood cells) to hold onto more oxygen and to stop red blood cells from becoming misshapen.
At that time, activity around the disease accelerated.
As Gina Kolata, one of the US reporters most interested in the disease pointed out, in 2018 “the National Institutes of Health was starting a program called the Cure Sickle Cell Initiative, meant to spur the development of gene therapies. A biotech company, Bluebird, was in the earliest phase of a gene therapy trial, hoping to develop a cure. So was Boston Children’s Hospital, which was beginning its own gene therapy trial. Then the biotech companies Vertex and CRISPR Therapeutics jumped in with a trial of their own.”
These latest therapies are based on gene editing. And so we come to 2023.
The arrival of CRISPR gene editing
We have known since 1949 that sickle cell disease is a molecular disease. So, it is perhaps not surprising that a new discovery popularly known as ‘molecular scissors’ would be applied to this disease – the real surprise is however how fast this happened.
‘Molecular scissors’ were discovered in around 2012, more technically known as CRISPR-Cas9, short for Clustered Regularly Interspaced Short Palindromic Repeats and CRISPR-associated protein 9. CRISPR is a method used by bacteria to snip out viruses in order to get rid of them. Now it is used to cut and edit DNA – also called gene or genome editing. In 2020 Jennifer Doudna and Emmanuelle Charpentier received the Nobel Prize for the discovery of CRISPR.
Since 2012 CRISPR has mainly been used for research and development, not as a treatment. Now it has become a type of therapy. First attempts at using it for sickle cell were made in 2017.
In March 2023 a Genome Summit was held in London, in which the potential of gene editing for treating inherited diseases was discussed and one of the first patients, Victoria Gray, involved in the gene therapy trial for the treatment of sickle cell disease gave a speech, highlighting the hopes and challenges of that treatment. She became something of a celebrity patient voice.
MHRA and FDA announcements
On 16 November 2023, the UK’s Medicines and Healthcare products Regulatory Agency (MHRA) announced the approval of a therapy based on gene editing for the treatment of sickle cell disease as well as for β-thalassaemia (a blood disorder that reduces the production of haemoglobin).
“On October 31, an FDA Advisory Committee panel met to discuss the risks and benefits of the investigational treatment ‘exa-cel’ (exagamglogene autotemcel) from CRISPR Therapeutics (CRSP) and its ‘big brother’ partner Vertex Pharmaceuticals (VRTX).” The gene therapy exa-cel is sold under the brand name Casgevy. CRISPR Therapeutics was founded by Emmanuelle Charpentier.
On 8 December the FDA announced its approval of two treatments for sickle cell disease, one using ‘Casgevy’, also approved by the MHRA, the other Lygenia (lovotibeglogene autotemcel or lovo-cel). “Casgevy is the first treatment to be approved that uses CRISPR. Patients will also need expensive, intensive medical care and a long hospitalization. The other treatment, called Lyfgenia and made by Bluebird Bio of Somerville, Mass., uses a common gene therapy method to add a good hemoglobin gene to patients’ DNA.” It is not based on gene editing.
Both the MHRA and the FDA seem to have deemed potential off-target effects (unintended genetic modifications) of clinical genome editing as low risk.
How does the therapy based on gene editing work?
An article in Nature explains: “Exa-cel aims to trigger production of a form of haemoglobin that is normally made only in developing fetuses. Production of this fetal haemoglobin is typically shut off soon after birth by a gene called BCL11A. Exa-cel disables BCL11A, allowing fetal-haemoglobin production to resume. This provides some haemoglobin that is not misshapen, and dampens the effects of the abnormal form.”
How is it done?
Doctors remove cells from each patient’s own (not a donor’s) bone marrow, edit a gene with CRISPR and then infuse billions of the modified cells back into patients. The edited cells produce a form of haemoglobin known as fetal haemoglobin, restoring normal red blood cell function.
The treatment is not simple and involves chemotherapy and long stays in hospital. But, in principle, after successful treatment, patients should no longer need to go to hospital for the treatment of pain crises or for blood transfusions, and so on.
What does it mean for people getting the treatment?
In the the post on identity, I’ll explore what such a radical treatment means for people getting it, not only in terms of becoming healthier, but also in terms of becoming somebody they haven’t been before.
Image: Wikimedia Commons: Normal blood cells next to a sickle blood cell, coloured scanning electron microscope image
No comments yet, fill out a comment to be the first





{kind=link}
Leave a Reply